
What Is Docking ? An In-Depth Scientific Overview of Molecular Docking
23rd Mar 2026
What Is Docking ? An In-Depth Scientific Overview of Molecular Docking
Introduction
In the realms of computational biology and drug discovery, understanding molecular interactions at the atomic level is crucial. This is where docking comes into play. But what is docking, and why has molecular docking become a cornerstone technique in modern pharmaceutical and structural biology research?
Docking provides a computational approach to predict how molecules, such as small ligands and macromolecules, interact. These predictions help guide structure-based drug design, enzyme engineering, and biomolecular interaction studies, saving time and resources in experimental workflows.
What Is Docking?
Docking is a computational technique that predicts the preferred orientation of one molecule (the ligand) when bound to another molecule (the receptor), forming a stable complex. The method simulates molecular recognition processes, which are central to biological functions such as:
- Enzyme-substrate binding
- Receptor-ligand signaling
- Antibody-antigen interactions
The purpose of docking is twofold:
- Predict Binding Poses – the 3D orientation of the ligand in the receptor binding site.
- Estimate Binding Affinity – a numerical measure of how strongly the ligand interacts with its target.
In silico docking serves as a predictive model for experimental studies, reducing the need for costly trial-and-error laboratory experiments.
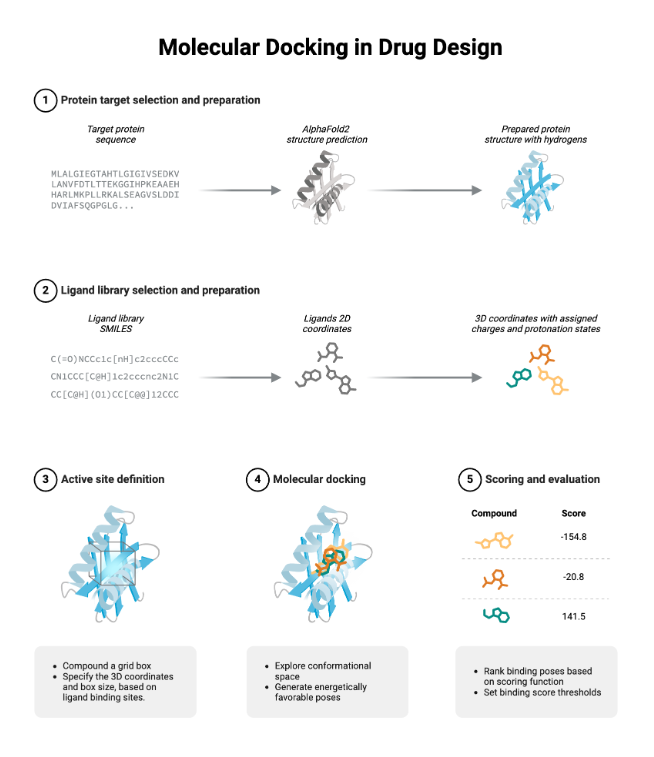
Molecular Docking: Definition and Scientific Basis
Molecular Docking is a specialized form of docking focused on ligand–macromolecule interactions, typically involving:
- Small molecule ligands (drugs, metabolites)
- Macromolecular targets such as proteins, DNA, or RNA
The primary goal of molecular docking is to identify the energetically most favorable complex between the ligand and its receptor.
This involves two critical steps:
- Conformational Search – exploring all possible orientations and conformations of the ligand in the binding site.
- Scoring Function Evaluation – ranking each pose using mathematical models that approximate the binding free energy.
Scientific Principles Behind Molecular Docking
1. Binding Models
Docking simulations are grounded in two classical models of molecular recognition:
- Lock-and-Key Model (Fischer, 1894): The ligand fits perfectly into a rigid receptor binding site.
- Induced Fit Model (Koshland, 1958): The receptor undergoes conformational changes to accommodate the ligand.
Modern molecular docking integrates flexible ligand docking and sometimes receptor flexibility to better mimic physiological conditions.
2. Scoring Functions
Scoring functions quantify the binding affinity between ligand and receptor. Types include:
- Force-field based – use classical mechanics to calculate interaction energies (e.g., van der Waals, electrostatic).
- Empirical – sum weighted contributions from hydrogen bonds, hydrophobic interactions, and desolvation.
- Knowledge-based – derive statistical potentials from known protein-ligand structures.
The accuracy of docking predictions depends heavily on the chosen scoring function.
3. Search Algorithms
Docking software uses computational algorithms to explore ligand conformations:
- Systematic methods – exhaustively sample ligand positions.
- Stochastic methods – random sampling using Monte Carlo or Genetic Algorithms.
- Simulation-based methods – integrate molecular dynamics to account for flexibility and solvent effects.
Efficient search algorithms are essential for large-scale virtual screening campaigns.
Types of Molecular Docking
Protein–Ligand Docking
The most common docking type, crucial for drug discovery. Predicts how small molecules interact with protein active sites.
Protein–Protein Docking
Used to study macromolecular interactions such as enzyme complexes or signaling pathways.
DNA/RNA–Ligand Docking
Analyzes interactions between ligands and nucleic acids, important in antiviral drug design and gene regulation studies.
Applications of Molecular Docking
1. Drug Discovery and Design
Molecular docking enables structure-based drug design by predicting ligand binding modes, guiding medicinal chemists to optimize drug candidates.
2. Virtual Screening
Computational screening of thousands to millions of compounds against a target receptor can rapidly identify potential hits for experimental validation.
3. Understanding Enzyme Mechanisms
Docking helps map substrate binding sites and analyze reaction mechanisms, essential for enzyme engineering.
4. Predicting Off-Target Effects
Docking can predict potential off-target interactions, improving drug safety profiles.
5. Personalized Medicine
Integration of docking with genomic data can predict patient-specific drug interactions.
Popular Molecular Docking Tools
Several software packages are widely used in research:
- AutoDock & AutoDock Vina – free, widely cited, versatile ligand and protein docking.
- Schrödinger Glide – commercial, high accuracy, integrates with molecular dynamics.
- MOE (Molecular Operating Environment) – comprehensive suite for drug design.
- DOCK – one of the first docking programs, optimized for protein-ligand interactions.
- SwissDock – web-based tool for academic users.
Each tool varies in its search algorithms, scoring functions, and handling of flexibility.
Advantages of Molecular Docking
- Cost-effective and faster than experimental approaches
- Enables virtual screening of large compound libraries
- Provides atomic-level insight into molecular interactions
- Supports rational drug design and lead optimization
Limitations of Docking
- Accuracy depends on protein structure quality
- Scoring functions may misrepresent real binding energies
- Limited treatment of receptor flexibility in some algorithms
- Does not fully account for solvent effects or entropic contributions
Despite these limitations, molecular docking remains a first-line in silico method before experimental validation.
Future Perspectives
The integration of machine learning, and molecular dynamics is revolutionizing docking. New trends include:
- Deep learning scoring functions for improved binding affinity prediction
- Enhanced flexibility models for more realistic simulations
- Integration with multi-omics data for precision medicine
Such advances promise faster, more accurate, and biologically relevant docking predictions, accelerating drug discovery and molecular research.
Conclusion
So, what is docking? At its core, it is a computational method that simulates molecular interactions. Molecular docking allows scientists to predict binding modes and affinities, guide drug discovery, and study biomolecular mechanisms in detail.
As computational power and algorithms advance, molecular docking is becoming an essential tool for modern biology and pharmaceutical research.
References
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A. 1958;44(2):98–104.
- Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte der deutschen chemischen Gesellschaft. 1894;27(3):2985–2993.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking. J Comput Chem. 2010;31(2):455–461.
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: A powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157.